Nicole H. Purcell, PhD, leads the Cardiovascular Signaling Laboratory at Huntington Medical Research Institutes (HMRI). Dr. Purcell completed her PhD in Molecular and Cellular Pathology at the University of Alabama Birmingham, her postdoctoral work with Dr. Jeffery Molkentin at Cincinnati Children’s Hospital Medical Center, and later at the University of California San Diego with Dr. Joan Heller Brown. She was appointed an Assistant Professor in the Department of Pharmacology in 2011 and an Associate Professor in 2017. In 2021, she moved her laboratory to HMRI and became an Associate Professor of Cardiovascular Research and Scientific Director of the Education Programs.
Dr. Purcell’s long-standing research interests have focused on delineating the multifactorial mechanisms that underlie signaling pathways associated with cardiac hypertrophy, heart failure, and ischemic brain injury. In particular, the Cardiovascular Signaling Lab focuses on the role of serine/threonine phosphatases (PHLPP) and AGC kinases in cardiovascular disease and aging. The goal is to advance the current understanding of disease etiology and develop mechanistic-based therapies beyond surgical intervention.
The area of research on PHLPP is in its infancy and the regulation of the PHLPP isoforms and their downstream targets are just beginning to emerge in cellular studies. In humans, the levels of PHLPP isoform expression are altered in patients with cancer, diabetes, and obesity, all of which are associated with aging (Figs. 1, 2).
PHLPP and Disease
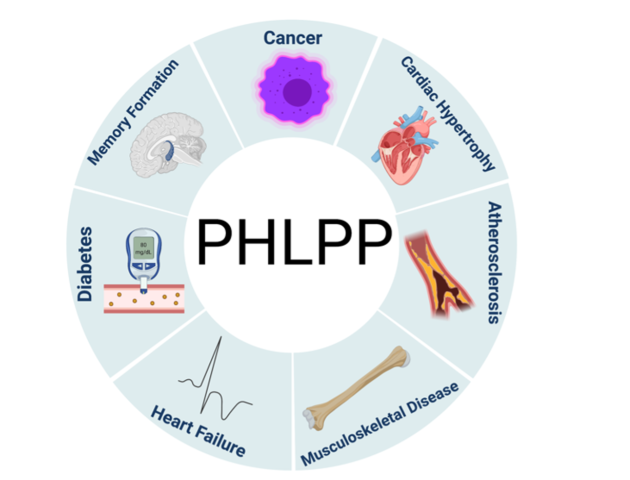
Fig. 1 Initially found as a tumor suppressor to play a key role in cancer, dysregulation of PHLPP isoforms has since been discovered to be involved in various pathophysiological states. PHLPP has been linked to several diseases, including cancer, metabolic disorders (diabetes), brain injury and memory formation, cardiovascular disease (cardiac hypertrophy and heart failure), musculoskeletal disease, and atherosclerosis. Modified from Lemoine et al 2021.
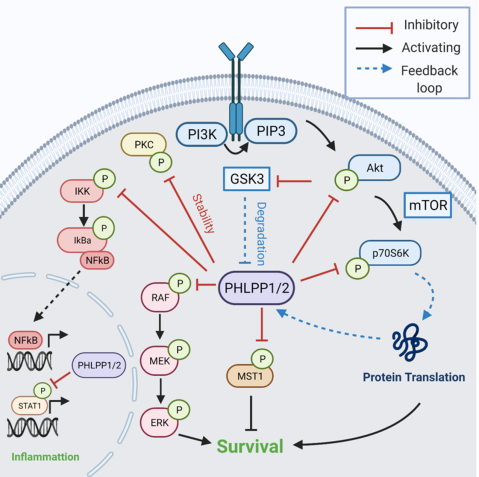
Fig. 2 PHLPP Signaling The PHLPP isoforms are tumor suppressors in cancer and inhibit cell growth and survival. The first identified substrate of PHLPP was the AGC kinase Akt, in which PHLPP selectively dephosphorylates the Akt isoforms on its hydrophobic motif. Additional PHLPP targets include Protein Kinase C (PKC), Macrophage Stimulating Protein 1 (MST1), p70S6 kinase 1 (p70S6K), and the RAF/MEK/ERK cascade, which PHLPP dephosphorylates to inhibit cell survival. Additionally, PHLPP dephosphorylates Signal Transducer and Activator of Transcription 1 (STAT1) and Inhibitor of Nuclear Factor Kappa B Kinase Subunit Beta (IKKB) to regulate inflammation. Two negative feedback loops regulate PHLPP expression. High levels of PHLPP expression reduce p70S6K activity and therefore reduce PHLPP protein translation and expression. Additionally, increased PHLPP levels reduces Akt activity and releases the inhibitory brake on Glycogen Synthase Kinase 3 (GSK3), leading to phosphorylation and degradation by the proteasome. Modified from Lemoine et al 2021.
Cardiac Hypertrophy
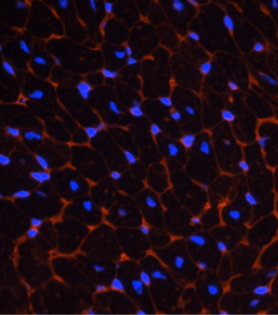
The laboratory has found that a newly discovered phosphatase, PHLPP, negatively regulates Akt activity and survival in the heart. Dr. Purcell’s research focuses on further understanding through the removal of PHLPP, its isoform specificity for Akt and target substrates following cardiac injury and determining the potential for PHLPP as a target for therapeutic strategies to improve cardiac function. Studies in this area use in vitro cell culture systems (i.e., NRVM and AMVM) and in vivo transgenic and knockout mouse models for phenotypic and morphometric analysis of cardiac hypertrophy and failure at baseline as well as after cardiac interventions (i.e., TAC, MI and I/R) (Fig. 3).
PHLPP1 Removal Accentuates Physiological Hypertrophy and Attenuates Pathological Hypertrophy
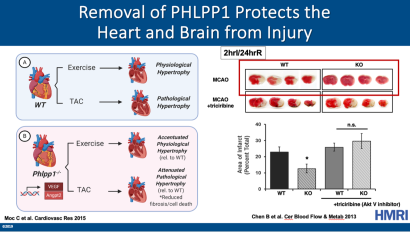
In wild-type (WT) mice, exercise induces physiological hypertrophy, whereas pressure overload via transverse aortic constriction (TAC) induces pathological hypertrophy, which leads to heart failure. (B) Removal of PHLPP1 increased Akt activity and capillary density in the heart and upregulation of angiogenic markers VEGF and Angpt2, without an increase in hypertrophy. Phlpp1-/- mice subjected to exercise showed accentuated physiological hypertrophy relative to WT mice as seen by an increase in heart size and myocyte cell area without hypertrophic gene expression or fibrosis. Following pressure overload (TAC), Phlpp1-/- mice had an attenuated pathological hypertrophic response relative to WT mice as demonstrated by reduced fibrosis, cell death, and preserved function. Modified from Lemoine et al. 2021. Following Middle Cerebral Artery Occlusion (MCAO), Phlpp1-/- mice had decreased ischemic injury (stroke) compared to WT mice as demonstrated by reduced infarct area in the brain. Inhibition of Akt activity with triciribine reduced the protection afforded with PHLPP1 removal. Increased Akt activity in the brain with PHLPP1 removal protects against ischemic stroke. Modified from Chen et al. 2013.
Neuroinflammation and the Aging Brain
A second area of interest in the laboratory investigates the effect of the PHLPP isoforms on the mechanisms regulating neuroinflammation and aging in the brain and the signaling pathways altered with nicotine exposure. The laboratory is currently investigating the pathogenic role of nicotine-induced inflammation on cognitive processes among the young. The brain is highly affected by oxidative stress and inflammation, which initiates intracellular signaling cascades that are both deleterious and beneficial to the brain. We have demonstrated that removing PHLPP1 increases Akt activity and protects the brain from ischemic damage following stroke (Chen et al. 2013) Fig. 4.Inhibition of Akt activity in the PHLPP1 knockout mice reverses the protection from ischemic damage. However, nothing is known in the brain about the regulation of PHLPP isoforms and the pathways it targets under stress conditions like aging and nicotine use (Fig. 5).
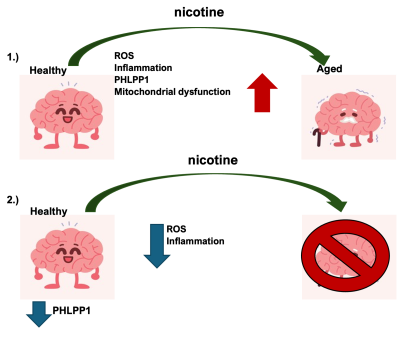
Fig. 5. Hypothesis Schematic: 1.) Exposure of healthy young brains to nicotine increases inflammation, oxidative stress, PHLPP1 expression, and mitochondrial damage, leading to an increased aging phenotype in the brain. 2.) Removal of PHLPP1 increases protective signaling pathways and reduces oxidative stress with nicotine exposure, thereby reducing the aging phenotype.
PHLPP gene-targeted mice and primary astrocytes are used to investigate novel mechanisms associated with signaling pathways leading to cell death and, in particular, the hypothesis that astrocyte viability is important to neuronal survival during ischemic injury (stroke) and nicotine use.
